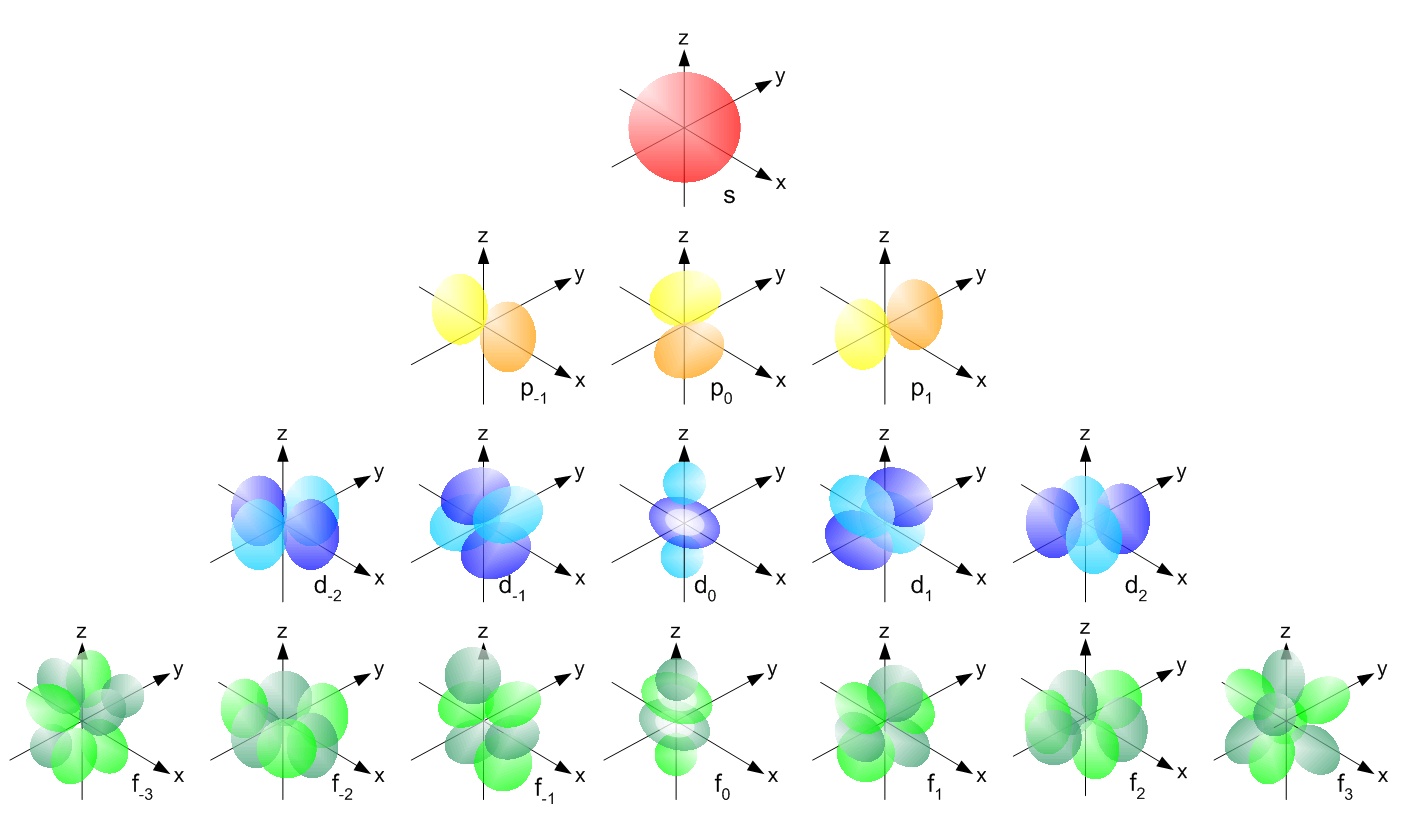
In the first post about atomic orbitals, it was mentioned that only atomic orbitals with the same symmetry can overlap to form molecular orbitals. For instance, s atomic orbitals form molecular orbitals with s atomic orbitals, px atomic orbitals with px atomic orbitals, and so on.
However, exceptions to this rule do exist.

MO diagram for nitrogen gas
Looking at the MO diagram above, Nitrogen gas obviously does not follow the rule because the 2s atomic orbitals are forming molecular orbitals with the 2p atomic orbitals. But how is this possible?? How come nitrogen gas can defy the basic rules of MO theory??
This is due to magical phenomena known as s-p mixing!
So, what is s-p mixing? This is a special process that only happen in elements where the s atomic orbital is very close in energy to the p atomic orbital. In this case, this usually happens to elements found before oxygen in the periodic table (hint: see nitrogen).

Diagram showing MO diagram before (a) and after (b) s-p mixing
The diagram above shows the MO diagram of a homonuclear diatomic compound before and after s-p mixing.
For S-P mixing to occur, the 2 interacting orbitals must be of the same symmetry and of similar energy. From the diagram above, we can see that the labelled orbitals are able to interact as they are of the same symmetry as labelled. Since they can interact, they must be of similar energy. A key point to note is that the greatest interaction occurs when the energy gap between the 2 interacting orbitals is the smallest. Such an interaction occurs between 2σg and 3σg as they are closest in energy.
Comparing (a) and (b), it is evident that s-p mixing lowers the energy level of the 2σ orbitals, thereby stablising them. At the same time, the 3σ orbitals are raised in energy. This way, the 3σ bonding orbital is higher in energy than the 1π bonding orbital. Thus following the Aufbau principle, the 1π bonding orbitals are filled before the 3σ bonding orbital. Thus, s-p mixing can change the magnetism of a compound.
One example of a compound that changes magnetism after s-p mixing is B2 as shown below:

MO diagram for B2 for no s-p mixing

MO diagram for B2 for s-p mixing
As seen, if s-p mixing occurs, B2 will be paramagnetic. This is because the electrons fill the 2π bonding orbitals singly before pairing up. Whereas in the non s-p mixing state, there is only one σ bonding orbital created by the p atomic orbitals and that means that the electrons pair up to fill the σ bonding orbital up completely. Therefore, diamagnetism is observed.
S-p mixing might not be prevalent, however, it can be useful to explain why some elements do not follow the expected trend of magnetism.