When the circuit breaker began back in April, research facilities across NTU were shut down and students and faculty alike were told to stay home.
For the most part, research activities were forced to a grinding halt.
But for SBS Assistant Professor Marek Mutwil, it was business as usual. In fact, the opposite of a research freeze occurred – so much data was generated that it sent the research team into a frenzy of activity, as they worked to prepare it for publication.
The paper in question? LSTrAP-Crowd: prediction of novel components of bacterial ribosomes with crowd-sourced analysis of RNA sequencing data. Helmed by Assistant Professor Mutwil, it also credits the staggering 285 undergraduate students of BS1009 Introduction to Computational Thinking. As part of a five-week-long project, the students were put to work processing terabytes of RNA-sequencing data.

Assistant Professor Mutwil at work, on a custom-built computer named Chuck Norris
There had been a great deal of uncertainty going into the project. After all, the program being used to process the data, LSTrAP-Crowd (Large-Scale Transcriptomic Analysis Pipeline in Crowd), had only been developed late-2019, just months before the start of the semester. It had barely been tested, and even then, by only seven users.
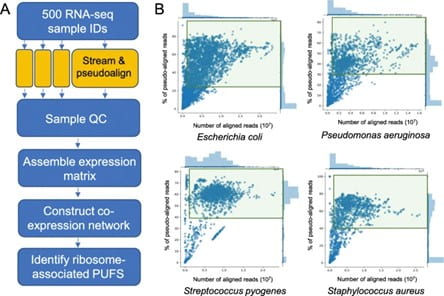
LSTrAP-Crowd pipeline and sample quality control
But when put into action, it went off without a hitch. Each student was armed with two CPU-cores and 12 GB of ram, courtesy of Google Colaboratory, a platform that LSTrAP-Crowd developer and BS1009 teaching assistant William Goh described as the “Google Docs for code”. The students were then split into groups, with each group assigned their own species of bacteria to download and process.
“That way, it gave the students a sense of ownership, and allowed their coding work to contribute directly to a real research paper,” William said.

BS1009 Examination
In the span of a single week, the 285 students of BS1009 had crunched through 26,269 gene expression samples from 17 of the most notorious bacterial pathogens – those that cause tuberculosis, pharyngitis, scarlet fever and so on – constituting a total of 26 terabytes of data.
To process a similar amount of data in the same timeframe, it would have required a supercomputer.
“It is a particularly impressive feat,” said Assistant Professor Mutwil, who is also the course coordinator for BS1009. “The students went from knowing nothing about programming, to producing a rather sophisticated software that can predict gene function.”
It was a win-win situation for all. According to BS1009 teaching assistant Tan Qiao Wen, the students progressed from knowing next to nothing about Python to solving problems on their own, and were interacting and analysing with real data just as researchers would.

Teaching Assistant Jonathan Ng tutoring a student during class
One student in particular stood out – Benedict Hew, a Biological Sciences undergraduate student, who is also the first author of the paper. “It was exciting to develop algorithms for something concrete – to help solve a problem,” he said.
With Benedict’s input, and working alongside Assistant Professor Mutwil, an analysis of the BS1009 project data was conducted. With the over-26,000 RNA sequences that had been processed, gene co-expression networks were constructed to identify novel genes involved bacterial protein synthesis.
The idea is that genes with similar expression profiles tend to be functionally related, which allowed the research team to draw connections and identify potential gene targets without any additional information. In the end, over a hundred previously uncharacterised genes involved in protein synthesis were identified, the results of which were later published in BMC Biology.
With its success, the project has opened up a whole new realm of possibilities for the field of antibiotic research. Given that growing antibiotic resistance has been projected to cause more deaths than cancer by 2050, the search for new antibiotics has never been more important. Here, the research has created a vital resource of potential antibiotic targets which can be used to uncover further vulnerabilities in bacteria and pave the way to developing more novel antibiotics.
As methods of analysis go, it has been a particularly unique (and effective) one. “Professor Mutwil did a really good job of coordinating everyone’s work online,” said Jonathan Ng, one of the teaching assistants for BS1009. In utilising crowd sourcing, the team has demonstrated how bioinformatics can be used as an alternative method of gene prediction, circumventing the usually cumbersome and labour-intensive approaches.
In the wake of a global pandemic, the project stands out not only for the prodigious number of people involved and the massive amounts of data processed, but also for highlighting the way research can adapt and carry on.

“Now, there is a lot of available biological data to analyse, and high-performance computing is readily available,” said Assistant Professor Mutwil. “I think only our imagination and inventiveness is the limit.”